Western Blot中文一般稱為蛋白質印跡。它是分子生物學、生物化學和免疫遺傳學中常用的一種實驗方法。其基本原理是通過特異性抗體對凝膠電泳處理過的細胞或生物組織樣品進行著色。通過分析著色的位置和著色深度獲得特蛋白質在所分析的細胞或組織中的表達情況的信息。
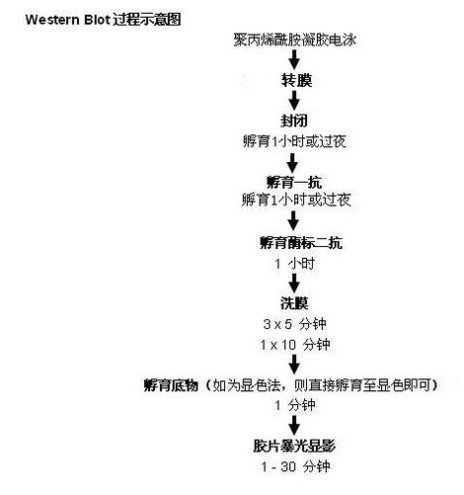
Western Blot與Southern印跡雜交或Northern印跡雜交方法類似,但Western Blot采用的是聚丙烯酰胺凝膠電泳 ,被檢測物是蛋白質,“探針”是抗體,“顯色”用標記的二抗。經過PAGE分離的蛋白質樣品,轉移到固相載體(例如硝酸纖維素膜NC膜 )上,固相載體以非共價鍵形式吸附蛋白質,且能保持電泳分離的多肽類型及其生物學活性不變。以固相載體上的蛋白質或多肽作為抗原,與對應的抗體起免疫反應,再與酶或同位素標記的第二抗體起反應,經過底物顯色或放射自顯影以檢測電泳分離的特異性目的基因表達的蛋白成分。該技術也廣泛應用于檢測蛋白水平的表達。其圖解為:
Western Blot顯色的方法主要有以下幾種: i. 放射自顯影 ii. 底物化學發光ECL iii. 底物熒光ECF iv. 底物DAB呈色。現常用的有底物化學發光ECL和底物DAB呈色,體同水平和實驗條件的是用**種方法,目前發表文章通常是用底物化學發光ECL。只要買現成的試劑盒就行,操作也比較簡單,原理如下(二抗用HRP標記):反應底物為過氧化物+魯米諾 ,如遇到HRP,即發光,可使膠片曝光,就可洗出條帶。
4. 蛋白樣本提取制備
蛋白樣品制備是Western Blotting的**步,更是決定WB成敗的關鍵步驟,總體原則和注意事項:
(1)盡可能提取完全或降低樣本復雜度只集中于提取目的蛋白(通過采用不同提取方法或選擇不同的試劑盒產品)
(2)保持蛋白的處于溶解狀態(通過裂解液的PH 鹽濃度 表面活性劑、還原劑等的選擇)
(3)提取過程防止蛋白降解、聚集、沉淀、修飾等,(低溫操作,加入合適的蛋白酶和磷酸酶抑制劑)
(4)盡量去除核酸,多糖,脂類等干擾分子(通過加入核酸酶或采取不同提取策略)
(5)樣品分裝,長期于-80 ℃中保存,避免反復凍融。
4.1細胞裂解
我們公司有針對細胞內的不同蛋白專門的蛋白裂解液試劑盒供您選擇。
4.11細胞裂解操作方法:
(1)培養的細胞經預冷的PBS漂洗2次,裂解液中加入蛋白酶和磷酸酶抑制劑。
(2)吸凈PBS,加入預冷的裂解液,((1 ml per 107 cells/100mm dish/150cm2 flask; 0.5ml per 5x106 cells/60mm dish/75cm2 flask).
(3)用細胞刮子刮取貼壁細胞,將細胞及裂解液溫和地轉移**預冷的微量離心管中,
(4)4℃搖動30 min。
(5)4℃離心12,000 rpm,10 min(根椐細胞種類不同調整離心力)
(6)輕輕吸取上清,轉移**新預冷的微量離心管中置于冰上,即為蛋白樣本,棄沉淀.
4.2組織裂解
(1)用滅菌的預冷的工具分離目的組織,盡量置于冰上以防蛋白酶水解,
(2)將組織塊放在圓底的微量離心管或Eppendorf管中,加入液氮凍結組織于冰上均質研磨,長期可保存于-80 °C。
(3)每約5 mg加入約300 μl 預冷的裂解液lysis buffer,冰浴勻漿后置于4℃搖動2小時,裂解液體積與組織樣本量有適當比例,(**終的蛋白濃度**少達到0.1 mg/ml, 理想的蛋白濃度應為1-5 mg/ml).
(4)4℃離心12,000 rpm,20 min,
輕輕吸取上清,轉移**新預冷的微量離心管中置于冰上,即為蛋白樣本,棄沉淀。
4.3蛋白酶和磷酸酶抑制劑
推薦購商品化蛋白酶和磷酸酶抑制劑復合試劑盒或COOKTAIL,或按下表配制:
備注:其中Sodium orthovanadate配制活化方法如下:
所有步聚均需在通風櫥中進行:
(1) 用雙蒸水配制100 mM 正礬酸鈉溶液.
(2)用鹽酸HCl 調**pH 9.0
(3)煮沸**溶液無色,盡量減少水分揮發.
(4)冷卻**室溫
(5)再調pH ** 9.0
(6)再煮沸**無色
(7)重復上述過程,直**溶液煮沸冷卻后達pH 9.0
(8)用水定容**原體積
(9)分裝保存于- 20 °C. 溶液變黃則棄之不用.
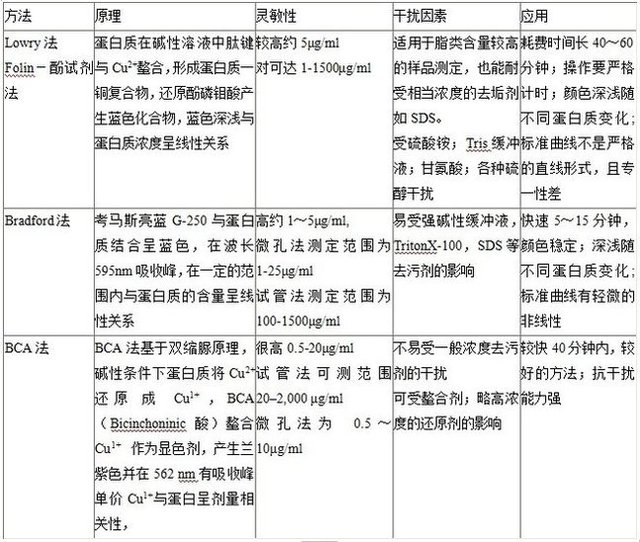
5. 蛋白定量
Bradford 法 Lowry 法或BCA 法 (操作簡單、需分光光度計或酶標儀),小牛血清白蛋白 (BSA) 作為標準曲線。如果裂解液中有NP40或其它表面活性劑,則推薦使用BCA 法。三種方法或產品比較
列表如下:
7. 電泳
7.1 PAGE膠的制備
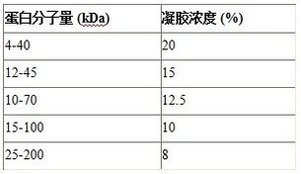
不同分子量的蛋白選擇不同的凝膠濃度(參考下表),原則上高分子量蛋白用低濃度膠,低分子量蛋白用高濃度膠分離。
不同濃度的凝膠的配制方見公司提夠的相關試劑盒說明書,首先配制各組分貯備液,然后分別配制濃縮膠和分離膠。
7.2蛋白分子量Marker
預染或非預染各種分子量的蛋白,用于標示電泳中蛋白的大小和示蹤
7.3陽性對照
目的蛋白或明確表達目的蛋白的組織或細胞的蛋白提取物,用于檢驗整個實驗體系和過程的正確性有效性/特別是一抗的質量和效率。建議使用該對照。可查閱文獻或抗體說明書選擇購買或自提該對照樣本。
7.4內參對照
管家基因編碼的、很多組織和細胞中都穩定表達的蛋白,用于檢測整個WB實驗過程及體系是否正常工作,并作為半定量檢測目的蛋白表達量的標準對照。必須設立。
7.5上樣與電泳
每孔上樣量為20-40 μg蛋白,使用專用槍頭或注射針頭,勿溢出加樣孔,電泳時間按電流儀說明書推薦方法使用,(1小時或過夜,取決于電壓大小),當染料到達膠的底部,關電源停止電泳,膠不能存放,應立刻進行下一步的轉膜。
8. 轉膜與顯色(Western Blot)
8.1膠中蛋白的檢測
電泳后檢測蛋白是否遷移正確與平均,可采用銅染或考馬斯藍染色檢測,如果凝膠中的蛋白需要進行轉膜則需可逆的銅染法,否則采用不可逆考馬斯藍法染色。
銅染法:電泳膠用蒸餾水洗數秒鐘,加入0.3 M CuCl2染色5-10分鐘,再用去離子水洗一次,在暗背景下觀察在蘭色膠背景下蛋白出現透明條帶,膠置于0.1- 0.25 M Tris/0.25 M EDTA pH 8.0緩沖液中漂洗脫色兩次,再置于電轉緩沖液中開始轉膜。
考馬斯藍法:用40%雙蒸水, 10% 醋酸, 50%甲醇的溶液固定膠中蛋白,考馬斯藍R-250染液(凱基產品)室溫染色4小時**過夜,保持搖勻,轉入67.5%雙蒸水, 7.5% 醋酸, 25%甲醇l搖勻**脫去多余的染料,蛋白被染成深藍色。
8.2蛋白轉膜
蛋白因結合SDS而帶電荷,在電場下從膠中轉**膜上,轉膜操作根椐電轉儀制造商的說明書進行轉膜方式分為半干和濕轉兩種,半干式轉膜速度快,而濕式成功率高并特別適合用于分子量大于100KD的蛋白。
濕式轉膜三明治排列為:海綿/紙/膠/膜/紙/海綿,全部緊密排列,特別是膠/膜之間不能留有氣泡,三明治安放的方向確認正確負極方為帶負電的膠里的蛋白,向正極方(膜)電遷移。標準的電轉緩沖液為1X Tris-glycine buffer 不含SDS,但加入20%甲醇,如果轉膜的蛋白分子量大于80KD,則推薦加入SDS使之終濃度為0.1%。
半干式轉膜中,三明治的排列為:/紙/膠/膜/紙,用電轉緩沖液浸濕后,直接置于電轉儀的正負極之間。膠于負極而膜置于正極。半干式的電轉緩沖液可不同于濕式的電轉緩沖液,推薦為:48 mM Tris, 39 mM glycine, 0.04% SDS, 20%甲醇,
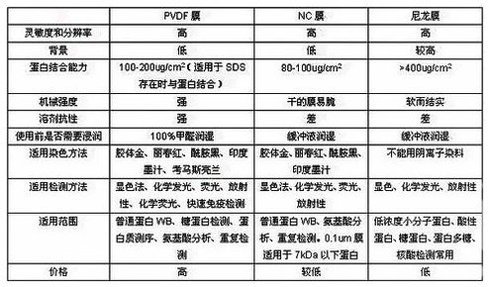
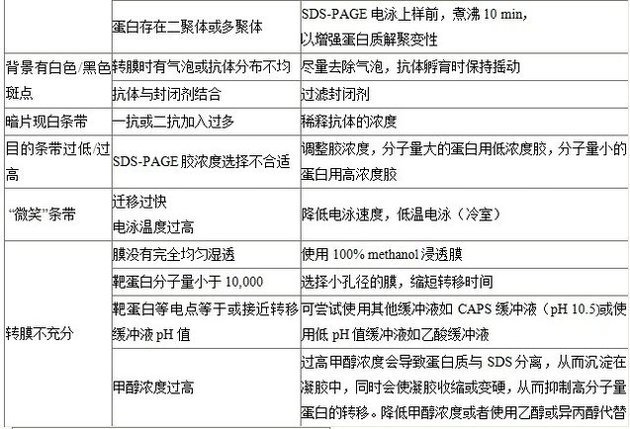
兩類膜可供選擇:硝酸纖維素膜和PVDF膜(正電荷尼龍膜),根據不同需要選擇(下表),PVDF膜需要浸泡甲醇中1-2分鐘,再孵育于冰冷的電轉緩沖液中5分鐘,膠也需在冰冷的電轉緩沖液中平衡3-5分鐘,否則轉膜時會導致條帶變形。
注:大蛋白和小蛋白的轉膜
電轉移緩沖液中SDS與甲醇的平衡、蛋白的大小、膠的濃度都會影響轉膜效果,如下調整可以增加轉膜效率:
大蛋白(大于100 KD)
1.對于大蛋白而言,其在凝膠電泳分離遷移較慢,而從凝膠轉出也非常慢,因此對于這種大分子量蛋白應該用低濃度的凝膠,8%或更低,但因低濃度的膠非常易碎,所以操作時需十分小心,
2.大蛋白易在凝膠里形成聚集沉淀,因此;轉膜時在電轉移緩沖液加入終濃度為0.1%的SDS,以避免出現這種情況,甲醇易便SDS從蛋白上脫失,因此應降低電轉移緩沖液中甲醇的濃度**10%或更低,以防止蛋白沉淀。
3.降低電轉移緩沖液中甲醇的比例以促進凝膠的膨脹,易于大蛋白的轉出。
4.如果使用PVDF,甲醇是必需的,且轉膜前PVDF需用甲醇活化。但如果是NC膜,甲醇可以不必加入電轉移緩沖液中。
5.選擇濕式,4 ℃轉膜過夜,以取代半干式轉膜。
小蛋白(小于100 KD)
1. SDS妨礙蛋白與膜的結合,特別是對小蛋白更是如此,因此,對于小分子的蛋白,電轉移緩沖液中可以不加SDS。
2.保持20%的甲醇濃度
對于大于500KD的蛋白,請參考下述文獻:Bolt and Mahoney, High-efficiency blotting of proteins of diverse sizes following sodium dodecyl sulfate–polyacrylamide, gel electrophoresis. Analytical Biochemistry 247,185–192 (1997).
更多的轉膜注意事項:
Ø避免用直接接觸膜,應使用鑷子,手指上的油脂與蛋白會封閉轉膜效率并易產生背景污斑
Ø排列三明治時,盡量用移液器或15ml試管趕除膠與膜之間的氣泡,或將三明治放在裝有的培養皿中以防止氣泡產生,請戴手套!
Ø確認裁剪的膜和濾紙與凝膠尺寸相同,否剛導致電流不能通過膜,從而轉膜無效
Ø雞抗體易于與PVDF膜和其它尼龍膜結合,導致高背景,請替換成硝酸纖維素膜以降低背景。
Ø膜上蛋白的檢測:麗春紅。為檢測轉膜是否成功,可用麗春紅染色,2%的麗春紅貯備液(20ML):: 2%麗春紅(0.4克)溶于30%三氯乙酸(6克)和30%磺基水楊酸(6克)麗春紅染色工作液:2%的麗春紅貯備液1:10稀釋,即加9 倍的ddH2O染色方法:將膜放入TBST洗一次,再置于麗春紅染色工作液中,在室溫下搖動染色5分鐘,大量的水洗膜直**水變清無色蛋白條帶清晰,(膜也可以用TBST或水重新洗后再進行染色)PVDF膜需用甲醇再活化后用TBST洗后進行封閉。
9. 膜的封閉
為防止一抗或/和二抗與膜的非特異性結合產生的高背景,因此需要進行膜的封閉,
傳統上有兩種封閉液:脫脂奶粉或BSA,脫脂奶粉成本低但不能用于磷酸化蛋白(因脫脂奶粉含有酪蛋白,該蛋白本身就是一種磷酸化蛋白),使用脫脂奶粉會結合磷酸化抗體從而易產生高背景。
某些抗體用BSA封閉時因不明原因可能會產生比脫脂奶粉更強的信號,,請仔細閱讀說明書注明的注意事項和膜的特殊的封閉方法。
配制5%脫脂奶粉或BSA 溶液:每100 ml TBST中加入5 g脫脂奶粉或BSA,混勻后過濾,如不過濾會導致使膜污染上細微黑顆粒。
封閉時,4°C搖動,封閉1 hour ,再用TBST洗5秒,進入下一步抗體的孵育。
10. 一抗的孵育
孵育Buffer:按抗體說明書建議的稀釋倍數,用TBST稀釋一抗,如果說明書沒有建議的稀釋倍數,則參照一般推薦的稀釋倍數(1:100-1:3000),一抗濃度過高會導致產生非特異性條帶。
某些實驗室傳統上在封閉液中孵育抗體,而有些實驗室用不含封閉劑的TBST來孵育抗體,結果因抗體而異,有時兩者結果相同,有時結果不同。
注:如果不存在高背景的問題,某些抗體用含低濃度(0.5 – 0.25%)脫脂奶粉或 BSA的封閉液來,稀釋,可產生相對更強的信號條帶。
孵育時間:一抗的孵育時間可從幾小時**過夜(一般不超過18小時)不等,取決于抗體與蛋白的親和性和蛋白的含量豐度,建議使用較高的抗體稀釋倍數和較長的孵育時間來保證特異性結合。
孵育溫度:盡可能低溫孵育,如果在封閉液中孵育一抗過夜,應在4oC進行否則會產生污染而破壞蛋白(降別是磷酸基團)。孵育一抗時需保持適當的搖動使之均勻覆沒膜,防止結合不均勻。
11. 二抗的孵育
一抗孵育結束后,用TBST搖動洗膜數次,每次5min或更長,去除殘留的一抗。
孵育Buffer和稀釋倍數:用TBST按說明書推薦的倍數稀釋二抗,如果說明書沒有標出稀釋倍數,則按常規的倍數稀釋(1:1000- 1:20,000)預試,二抗的濃度過高也會導致非特異性條帶。亦可以在封閉液中孵育二抗(和一抗),但可能在降低背景同時導致特異性條帶的信號也減弱,可能是封閉劑阻礙了抗體與靶蛋白的結合。
孵育時間和溫度:室溫、1-2 hours, 搖動。
二抗連接物:推薦使用二抗連接HRP,不建議連接AP堿性磷酸酶,因其不夠靈敏。
12. 顯色
顯色分為酶促底物發光和化學發光法或熒光法
酶促底物發光法代表為DAB顯色法,與其它同類方法的比較如下圖所示:
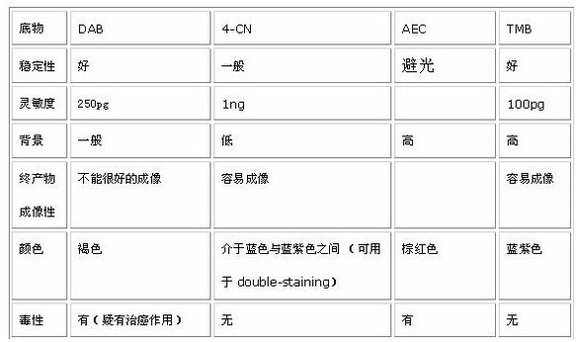
而現在**常用的是化學發光法:HRP化學發光底物 Luminol(ECL法chemiluminescence)及其改良法,
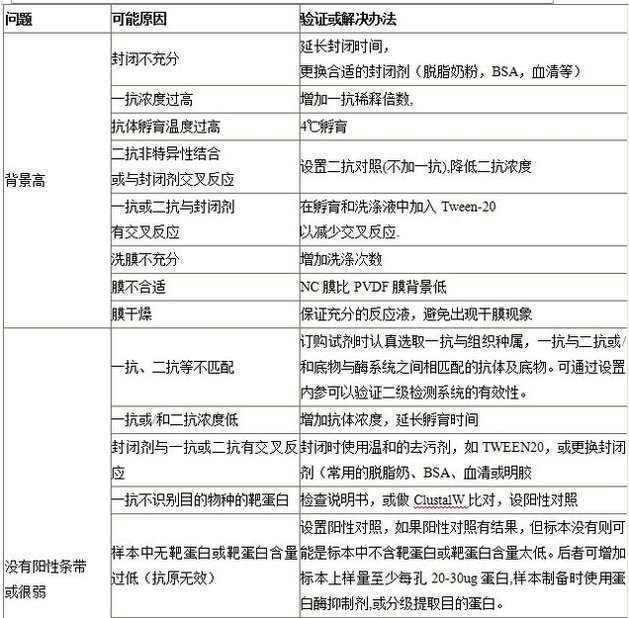
13. 常見問題分析與解決方案
13.2其他常見問題分析
無信號
一抗和二抗不匹配
二抗需和一抗宿主的物種相同(如一抗來自兔,二抗為抗兔抗體)。
沒有足夠的一抗或二抗結合目標蛋白
使用高濃度抗體。4°C延長孵育時間(如過夜)。
封閉劑與一抗或二抗有交叉反應
使用溫和的去污劑如吐溫-20,或更換封閉劑(常用的脫脂奶粉、BSA、血清或明膠)。
一抗不識別待檢物種的蛋白
參照說明書,比對免疫原序列和蛋白序列以確保抗體和目的蛋白會發生反應,設置陽性對照。
抗原量不足
每條泳道蛋白上樣量不低于20-30?μg,使用蛋白酶抑制劑并設置陽性對照。
組織中目的蛋白含量低
濃縮使信號**大化(例如檢測核蛋白可用核裂解物)。
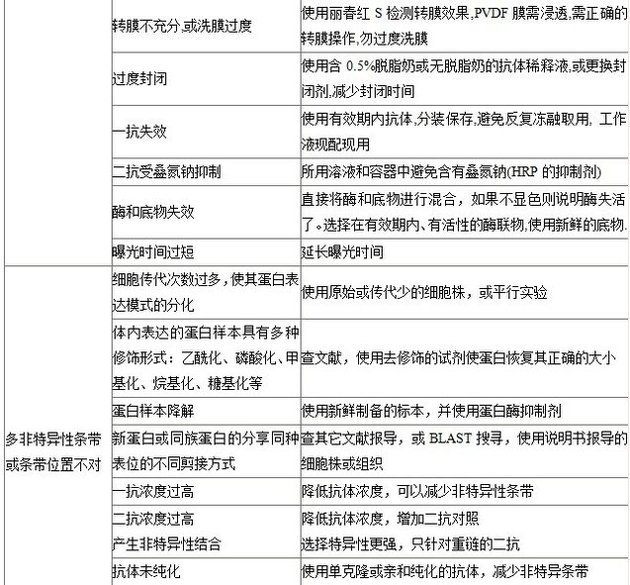
轉膜不充分
使用可逆染色劑如麗春紅S?檢測轉膜效果,檢查轉膜操作是否正確。PVDF?膜需預先浸在甲醇中,然后浸到轉移緩沖液中。
洗膜過度
勿過度洗膜。
過度封閉使目標蛋白不能顯色
使用0.5%奶粉或無奶粉代替5%奶粉的抗體稀釋液,或更換封閉劑,減少封閉時間。
一抗失效
使用新鮮抗體,重復使用有效濃度會降低。
二抗受疊氮鈉抑制
避免疊氮鈉和HRP?標記抗體一起使用。
檢測試劑盒過期和底物失活
使用新鮮的底物。
背景高
未進行非特異性封閉或封閉不充分
延長封閉時間,考慮更換合適的封閉劑。Abcam推薦5%脫脂奶粉、3% BSA或血清封閉30?分鐘,這些可以包含在抗體緩沖液中。
一抗濃度過高
稀釋抗體**合適濃度,以更高稀釋度抗體延長孵育時間(耗時長但特異性結合**好)。
抗體孵育溫度過高
4°C孵育。
二抗與封閉劑非特異性結合或反應
設置二抗對照(不加一抗)。
二抗與封閉劑非特異性結合或與封閉劑反應
在孵育和洗滌液中加入溫和去污劑如吐溫-20。脫脂奶粉含有酪蛋白,該蛋白本身就是一種磷酸化蛋白,會結合磷酸化特異性抗體而易產
生高背景。使用BSA代替奶粉作為封閉劑。
未結合蛋白質洗滌不充分
增加洗滌次數。
選膜不當產生的高背景
硝酸纖維素膜比PVDF膜背景低。
膜干燥
在孵育過程中防止膜變干,每一步都要保證膜有充分的反應液,可放入攪拌子不斷攪動或輕輕振蕩使膜浸在溶液里。
多帶現象
細胞傳代次數過多,蛋白表達不同
使用原始未傳代的細胞株,和現在的細胞株一起做平行對照實驗。
體內表達的蛋白樣本具有多種修飾形式如乙酰化、甲基化、烷基化、磷酸化、糖基化等
參考文獻,使用試劑使樣品去磷酸化、去糖基化來證明翻譯后的修飾。
蛋白樣本降解(蛋白質分子量降低)
在樣品緩沖液中加入足夠的蛋白酶抑制劑。
檢測到未經報道過的新蛋白或同一蛋白家族中具有相似表位而結構不同的蛋白
查閱其它文獻報道,或BLAST搜尋,使用說明書推薦的細胞株或組織。
一抗濃度過高,高濃度時常出現多條蛋白帶
降低抗體濃度和/或孵育時間。
二抗濃度過高,高濃度產生非特異性結合
降低抗體濃度,加入二抗對照(不加一抗)。
抗體未經純化
使用親和純化的抗體,減少非特異條帶。
條帶為非特異性條帶
應用封閉多肽來區分特異性和非特異性條帶,只有特異性條帶能被封閉從而消失。
目標蛋白形成多聚體
SDS-PAGE電泳加樣前,煮沸10分鐘而不是5分鐘,使蛋白質解聚。
背景有不均勻的白色斑點
轉膜時膜上有氣泡或抗體在膜上分布不均
轉膜過程中盡量除去氣泡,抗體孵育時保持搖動。
背景有黑色斑點
抗體結合了封閉劑
過濾封閉劑。
深背景出現白色條帶
一抗或二抗加入過多
稀釋抗體的濃度。
分子量標準條帶呈黑色
抗體和分子量標準發生了反應
在分子量標準和**個樣品之間增加一個空白條帶。
目的條帶染色過低/過高
分離不徹底
改變凝膠比例:分子量大的蛋白用低濃度膠,分子量小的蛋白用高濃度膠。
條帶“微笑”效應
1.遷移過快
2.電泳溫度過高(改變了pH值和遷移速度)
降低遷移速度或低溫電泳(冷庫或冰上)。
相同的蛋白雜交出現大小不均勻條帶
制備凝膠時凝膠凝固太快,致使泳道中丙烯酰胺的比例不均勻
參照凝膠的配方,在凝膠中加入適量TEMED,放置時在凝膠頂部加入適量0.1% SDS(水稀釋)以防凝膠變干。
凝膠染色不均勻
1.細菌污染
2.抗體量不足
(1)4 °C保存抗體并使用新鮮的緩沖液浸泡凝膠。
(2)確保振蕩條件下孵育膜或抗體充分覆蓋膜。